

May 23, 2017
- From the desk of Nigel R. A. Beeley, Ph.D., CDD Advocate -
The chemical database of today is inextricably linked to several historical threads, not all of them chemical, that began a long time before the modern computational world existed. Understanding these origins provides a deeper understanding of the modern chemical database and yields perspective on the paths that brought us here.
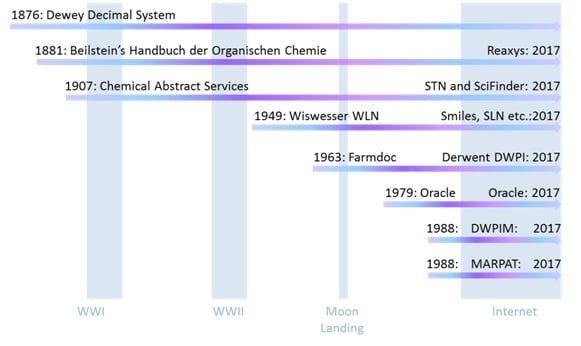
The library thread began in 1876, independent of any chemical specific factors, with the Dewey Decimal Classification, along with the use of card indexing (1), which undoubtedly made others think about how best to organize and catalog data. It survives to this day in the remaining library facilities that still have books and papers. The sciences are all in the 500 grouping (540: chemistry, 570: biology) (2). Already at that time chemists were publishing papers containing information about significant numbers of molecules and the importance of curating and managing that information had occurred to several. Beilstein’s Handbuch der Organischen Chemie (3) began life in 1881, became computational in the late 1970’s, and today is a fully searchable package called Reaxys. Chemical Abstracts Services (CAS) started in 1907 (4), also became computational in the 1970’s and today consists of two fully searchable packages, SciFinder and STN. The Derwent World Patents Index (DWPI) began life as Farmdoc in 1963 (5), coinciding with a worldwide expansion of Pharmaceutical Research, and at the time was the only organization to have thought through how to handle the genera of structures that so often appear in the chemical patent literature, the so-called “Markush” chemical structures, in a searchable way. US Patent 1506316 was the first issued patent to include a genus of related chemical structures, subsequently named “Markush” structures after the inventor of this patent, Eugene A Markush (6). The history of the DWPI is illustrative of how important being compatible to the legacy of manual abstracting and record keeping has been as these organizations transitioned from a paper world to a computational world. Thus in 1987 their original abstracting method had to be complemented with “Markush TOPFRAG” software to automatically convert a variable structure drawn by the user into the correct chemical code search strategy (7). In order to better take advantage of developments in relational databases Derwent introduced the fully searchable DWPIM in 1988, but did not include any historical data. So the two databases coexisted until 2005 as a source of active patent documents, based on the minimum patent life of 17 years at the time (8). CAS also introduced their own computational database of Markush structures called MARPAT in 1988 (9). It should also be pointed out that tracking down data and keeping up with the literature prior to the computational era was an arduous task, involving CAS, Beilstein, hundreds of published journals and hand filled card indexes and it wasn’t unusual for scientists to spend more time in the library than actually doing experiments, not even including the armies of professional library search staff, patent experts and nomenclature specialists etc. employed for support.
The skeletal chemical formulae thread that we are so familiar with today began with Kekulé (10) and his theoretical depiction of the structure of benzene, followed by Wilhelm van Hoffman who introduced molecular models, albeit as a 2D representation of structure (11). Then Van't Hoff and Le Bel (12) independently suggested organic molecule representations can be three-dimensional. Molecular models began to assume their modern appearance, and the depiction of organic molecules as “chicken wire” on paper which could be hand drawn began in earnest. Today there are rules and conventions from IUPAC for drawing chemical structures (13).

Young and enthusiastic colleagues (Sylvia Ernst, now with CDD, is in the center), manually curating chemical reaction data in the pre-computational era of Beilstein (photo courtesy of Sylvia Ernst).
The computer representation of chemical structure is an important thread. Given the diverse backgrounds and objectives of the traditional paper database organizations, and their penchant for doing their own thing (CAS and IUPAC still use different nomenclature conventions to this day) (14) there were differences in approaches to the computational depiction of chemical structures. The crystallographers were the first to get involved. The theoretical principles of crystallography had been described in the late 1800’s (15) but could not be reduced to practice until electromagnetic radiation sources of suitable wavelength, in the form of X-rays became available. In 1913 William Lawrence Bragg (the youngest ever Nobel laureate in Physics at age 25) introduced the modern era of crystallography by solving the structures of sodium chloride and diamond using X-ray crystallography (16). Crystal coordinates and the related cartesian coordinates derived from X-ray crystallographic experiments became important data sets which represented chemical structures in 3-dimensional space. The protein crystallographers expanded on this. The first attempts by crystallographers at displaying protein structures on a cathode ray tube (17), the first iterations of the Protein Data Bank in 1969 (18) and the ORTEP plotters of the early 1970’s (19) all used cartesian coordinates. Molecular mechanics experts joined the groups of crystallographers who began using IBM punch cards to perform theoretical calculations on molecules where inputs and outputs were sets of cartesian coordinates (20). Cartesian coordinates also had the advantage that there were standard math solutions buried in graph theory that allowed one set of coordinates to be compared with another (21). Not surprisingly, therefore, when MDL burst on the scene in the late 70’s, their standard molfile looked like a set of two dimensional cartesian coordinates with a connectivity table and other brief instructions such as atom type and connectivity contained within. Today this has evolved into the still widely used sd file format for collections and libraries of compounds (22).
But all was not well. These files of cartesian coordinates were not easily searchable, not scaleable to millions of compounds and, furthermore, were not easily adaptable to relational database technologies such as Oracle, which were rapidly becoming the gold standard in general purpose searchable databases. Those of us who recall working in Pharmaceutical R&D in the 1980’s will remember how poor MDL’s MACCS system was at handling any sort of data other than chemical structures. Hence the arrival of new representations, or the revival of the not so new in the form of the line/string notations thread. These began with the Wisswesser line notation (WLN) in 1949 (23). It was the basis of ICI Ltd's CROSSBOW database system developed in the late 1960s (24) and was also the tool used to develop the CAOCI (Commercially Available Organic Chemical Intermediates) database, the datafile from which Biovias' ACD file was developed (25). WLN is still being extensively used by BARK Information Services. It originated at a time when researchers were unaware of the term “user friendly” and it was not intuitive to the chemist. The more chemist-friendly Smiles strings came along from David Wieninger in 1988 (26), about the same time that combinatorial chemistry was getting going and the prospect of actually synthesizing and testing full sets of “Markush” structures became real (27). Variants in Smiles came along later and included SMARTS (28), Tripos Inc.’s SLN (29), and Beilstein’s ROSDAL (30). Even IUPAC got into the game with InChi in 2006 (31). Open SMILES was established in 2007, perhaps as a response to InChi (32).
All of these line/string notations provided the tools to integrate chemical databases with other sources of data but didn’t actually complete the job. The final thread was the introduction of the Oracle cartridge (33) which provided the much needed interface to generating Structured Query Language (SQL’s) to interrogate both chemical structures held in line notation format and other alphanumeric data such as test results, and it was amenable to people writing suitable “cartridge” software. It is also important to note that the various line notations provided easy ways to perform sub-structure and similarity (Tanimoto coefficient) searching (34) as well as methods to assign key parameters such as numbers of rotatable bonds (important for Lipinski’s rule of 5) (35).
This is an ongoing process even today, but considerable progress has been made. The modern architecture for a useful database is a series of computational layers. At the top is a graphical user interface, typically web based, which allows the input of chemical structures and substructures for search queries, as well as conventional text and numerical queries with some elements of Boolean logic. Then there are layers of software which convert the structural and textual query into something suitable for relational database interrogation, usually in the form of Structured Query Language (SQL) for Oracle or MySQL. The database is then queried in a relational way and the answers are returned to be processed again by the intermediate layers of software which, in turn, deliver chemical structures and textual answers which are displayed to the scientist, all in a fraction of a second.
Although the history tracing the origins of chemical databases is fascinating, as are the details regarding how things work, practicing pharmaceutical R&D scientists today need know little of this and can focus exclusively on asking the databases questions and getting answers back instantaneously, using software and database management services such as CDD Vault (36), rather than spending weeks buried in paper files and libraries, as did previous generations of researchers. The impacts on productivity are truly staggering. Investigation of synthetic pathways, numbers of compounds synthesized and tested, analysis of results and SARs, patentability, curation of data, report writing, and preparation of FDA documents can all done in a fraction of the time and with far fewer support staff when compared to the world prior to “chemoinformatics” (37).
References:
1) https://en.wikipedia.org/wiki/Dewey_Decimal_Classification
2) https://en.wikipedia.org/wiki/List_of_Dewey_Decimal_classes
3) https://en.m.wikipedia.org/wiki/Beilstein_database
4) https://www.cas.org/
https://en.m.wikipedia.org/wiki/Chemical_Abstracts_Service
5) https://en.m.wikipedia.org/wiki/Derwent_World_Patents_Index
6) https://en.m.wikipedia.org/wiki/Markush_structure
US 1506316 (1924) “Pyrazolone dye and process of making the same”. Eugene A Markush
7) http://www.stn-international.com/uploads/tx_ptgsarelatedfiles/piug1.pdf
The Complete Markush Structure Search: Mission Impossible ? (2001)
8) http://www.stn-international.de/uploads/tx_ptgsarelatedfiles/DCR_and_DWPIM_seminar_20160519_01.pdf
Comprehensive DWPISM structure searching using DCR and DWPIM on STN (2016)
9) http://www.cas.org/content/markush
10) https://en.wikipedia.org/wiki/August_Kekul%C3%A9
11) https://en.wikipedia.org/wiki/August_Wilhelm_von_Hofmann#Molecular_models
12) https://en.wikipedia.org/wiki/Jacobus_Henricus_van_%27t_Hoff
https://en.wikipedia.org/wiki/Joseph_Achille_Le_Bel
https://en.wikipedia.org/wiki/Le_Bel%E2%80%93van%27t_Hoff_rule
13) Jonathan Brecher (2006) “Graphical representation of stereochemical configuration (IUPAC recommendations 2006)” Pure Appl. Chem., 78 (10): 1897–1970
14) https://en.wikipedia.org/wiki/IUPAC_nomenclature_of_organic_chemistry
Naming and Indexing of Chemical Substances for Chemical Abstracts (2007)
15) https://en.wikipedia.org/wiki/X-ray_crystallography
16) https://en.wikipedia.org/wiki/William_Lawrence_Bragg
17) Generally attributed to Leventhal and Langridge (1966) Eric Francoeur (2002) “Cyrus Levinthal, the Kluge and the origins of interactive molecular graphics”. Endeavour 26 (4) 127-1312002
18) https://en.wikipedia.org/wiki/Protein_Data_Bank#History
19) https://en.wikipedia.org/wiki/Molecular_graphics
20) https://en.wikipedia.org/wiki/Cartesian_coordinate_system
21) https://en.wikipedia.org/wiki/Graph_theory
22) https://en.wikipedia.org/wiki/Chemical_table_file#SDF
23) William J. Wiswesser (1982). "How the WLN began in 1949 and how it might be in 1999". J. Chem. Inf. Comput. Sci. 22 (2): 88–93
24) https://en.wikipedia.org/wiki/Wiswesser_line_notation
25) https://en.wikipedia.org/wiki/Accelrys
26) https://en.wikipedia.org/wiki/Simplified_molecular-input_line-entry_system
Weininger D (1988). "SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules". Journal of Chemical Information and Modeling. 28 (1): 31–6.
27) https://en.wikipedia.org/wiki/Combinatorial_chemistry
28) http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html
29) https://en.wikipedia.org/wiki/SYBYL_line_notation
30) https://books.google.com/books?id=ZV_yCAAAQBAJ&pg=PA179
31) https://en.wikipedia.org/wiki/International_Chemical_Identifier
32) http://opensmiles.org/
33) https://en.wikipedia.org/wiki/SQL
https://docs.oracle.com/cd/B19306_01/appdev.102/b14289/dciwhatis.htm
34) https://en.wikipedia.org/wiki/Jaccard_index
35) https://en.wikipedia.org/wiki/Lipinski's_rule_of_five
36) https://www.collaborativedrug.com
37) https://en.wikipedia.org/wiki/Cheminformatics
F K Brown (1998). "Chapter 35. Chemoinformatics: What is it and How does it Impact Drug Discovery". Annual Reports in Medicinal Chemistry 33: 375-384
Further reading:
1) Introducing Cheminformatics by David Wild (2013). Available from Amazon as a Kindle E-book for $9.95
https://www.amazon.com/Introducing-Cheminformatics-David-Wild-ebook/dp/B00G5TS7B4/