

May 9, 2014
CDD Vault now allows you to build predictive models. Because we believe in collaboration and reproducible results, we are releasing the core algorithms that underly this feature as open source software. The chemical fingerprints are detailed below, and we have also released our modified Bayesian model. Send an email to info@collaborativedrug.com if you'd like to try using models in CDD Vault.
By Alex Clark; portions of this post originally appeared in Cheminformatics 2.0 As of now, the latest version of the popular open source Chemistry Development Kit (CDK) has its own implementation of the highly regarded ECFP and FCFP classes of chemical structure fingerprints (sometimes referred to as circular or Morgan fingerprints). While the general recipe for this kind of fingerprint has been available for awhile, and there are a number of implementations in various different toolkits, this one distinguishes itself in several ways: it has been implemented as closely as possible to the description of the original definition (without having access to the trade secrets that were left out of the paper); it includes resolution of chiral centers; it is freely available as open source Java code; and, last but not least, the algorithm is designed to be as portable as possible, with no major dependencies on specific programming languages or cheminformatics toolkits. [caption id="attachment_7271" align="alignnone" width="603"]
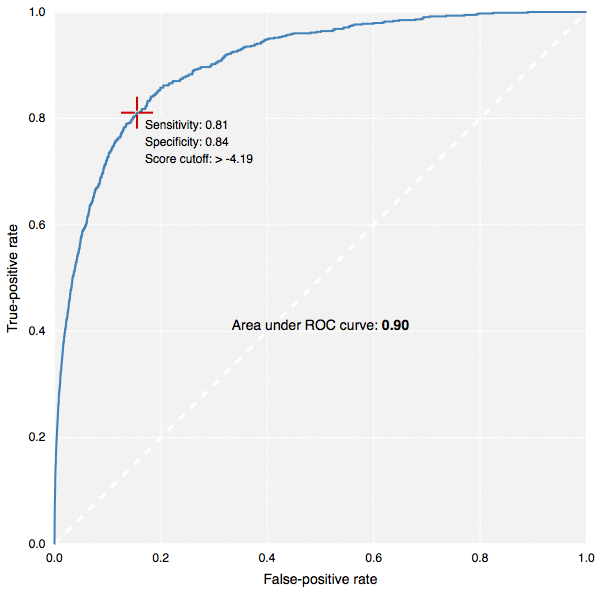 The new fingerprints in action: building predictive models in CDD Vault.[/caption] This contribution to CDK has been made by Collaborative Drug Discovery, and the implementation carried out by yours truly. It is used for the new predictive modeling features in CDD Vault, as well as the free TB Mobile app for tuberculosis research. Because the CDK version (available in our
Github fork, or in the latest & greatest main CDK branch), written in Java, produces fingerprints that are literally identical to the version that was coded up in Objective-C for use in iOS apps, it means that models can be created using a Java-based desktop application or webservice, and applied on the client side by the mobile app. This is how the TB Mobile app is able to provide similarity sorting, visual clustering and target activity prediction, all by mixing precalculated reference data with dynamically calculated user-supplied data. In case you’re not familiar with the terms ECFP6 and FCFP6, in a nutshell: the chemical structure is examined for all subgraphs with a diameter of up to size 6 (i.e. start with a single node, and do 3 breadth-first iterations). Each of these graphs is assigned a
hash code based on the properties of the atom, the bonds, and where applicable, chirality. These hash codes are put through several redundancy elimination steps, and eventually converted into a list of 32-bit integers. A druglike molecule typically has from dozens to hundreds of these unique hashcodes. Molecules that are structurally very similar tend to share a large number of these indices in common, and so are often compared using the Tanimoto coefficient. For ECFP-class fingerprints, the atom properties are somewhat literal (e.g. atomic number, charge, hydrogen count, etc.), whereas for the FCFP-class (“F” stands for
functional) the atom characteristics are swapped out for properties that relate to ligand binding (e.g. hydrogen donor/acceptor, polarity, aromaticity, etc.) which means that different atoms often start with the same value (e.g. -OH and -NH might be considered the same). There are many different types of graph-based fingerprints that can be used as alternative choices for various kinds of structural comparisons. The ECFP and FCFP categories have been used successfully in a number of studies, particularly for Bayesian model building. The way these fingerprints are constructed provides a good balance, giving empirically good proportionality when used for the various kinds of similarity comparisons, which has made them a popular choice for drug discovery. Multiple software vendors have implemented their own style of circular descriptors, but there exists a problem: the original invention is based on an algorithm that has been published in the literature, but unfortunately leaves out key details that make it not possible for anyone else to implement a version that is literally compatible. That may not matter if you are doing all of your modeling with software from a single vendor, but if you want to mix and match, fingerprints generated by one package cannot be compared to fingerprints generated by another, even if the input molecules are the same and the implementation follows the same basic recipe: the numbers will be completely different. Because the CDK project previously did not have its own implementation, we have filled this particular hole. Anyone using software in a Java runtime environment can have access to it without having to pay anyone or ask for permission. We have put in a significant amount of elbow-grease to make sure that these fingerprints pass various validation tests, and perform with an enrichment rate comparable to other implementations. But perhaps more importantly, the algorithm has been very deliberately built in a way that is relatively easy to describe in words, and is based on code that is highly self contained. Definitions like implicit hydrogen count, aromaticity, ring blocks and chirality are minimalistic, well defined, and guaranteed never to change. This means that if you generate a list of fingerprints for a structure, you can store them in a database, and use them forever; you don’t need to version them and make sure they get rebuilt whenever one of the dependencies changes (which is a major headache with many software packages). And because the implementation is quite platform agnostic, a single source file can be translated line-by-line into a different development environment. In practice, you can use the CDK implementation to generate sample results, to make sure that the transplanted version is operating identically. As mentioned previously, this has already been done and is in use by the
TB Mobile app. We intend to explicitly document the algorithm in the scientific literature in the near future, to complement the freely available source code, but you will have to wait for that. In the meanwhile, if you feel brave, look for the file
CircularFingerprint.java in the CDK source, under the fingerprints hierarchy. This is also the first time I have actively worked with the CDK codebase. The project appears to be in the midst of a major overhaul, so it will be interesting to see what comes out the other end. Besides an important new class of fingerprints, that is!
The new fingerprints in action: building predictive models in CDD Vault.[/caption] This contribution to CDK has been made by Collaborative Drug Discovery, and the implementation carried out by yours truly. It is used for the new predictive modeling features in CDD Vault, as well as the free TB Mobile app for tuberculosis research. Because the CDK version (available in our
Github fork, or in the latest & greatest main CDK branch), written in Java, produces fingerprints that are literally identical to the version that was coded up in Objective-C for use in iOS apps, it means that models can be created using a Java-based desktop application or webservice, and applied on the client side by the mobile app. This is how the TB Mobile app is able to provide similarity sorting, visual clustering and target activity prediction, all by mixing precalculated reference data with dynamically calculated user-supplied data. In case you’re not familiar with the terms ECFP6 and FCFP6, in a nutshell: the chemical structure is examined for all subgraphs with a diameter of up to size 6 (i.e. start with a single node, and do 3 breadth-first iterations). Each of these graphs is assigned a
hash code based on the properties of the atom, the bonds, and where applicable, chirality. These hash codes are put through several redundancy elimination steps, and eventually converted into a list of 32-bit integers. A druglike molecule typically has from dozens to hundreds of these unique hashcodes. Molecules that are structurally very similar tend to share a large number of these indices in common, and so are often compared using the Tanimoto coefficient. For ECFP-class fingerprints, the atom properties are somewhat literal (e.g. atomic number, charge, hydrogen count, etc.), whereas for the FCFP-class (“F” stands for
functional) the atom characteristics are swapped out for properties that relate to ligand binding (e.g. hydrogen donor/acceptor, polarity, aromaticity, etc.) which means that different atoms often start with the same value (e.g. -OH and -NH might be considered the same). There are many different types of graph-based fingerprints that can be used as alternative choices for various kinds of structural comparisons. The ECFP and FCFP categories have been used successfully in a number of studies, particularly for Bayesian model building. The way these fingerprints are constructed provides a good balance, giving empirically good proportionality when used for the various kinds of similarity comparisons, which has made them a popular choice for drug discovery. Multiple software vendors have implemented their own style of circular descriptors, but there exists a problem: the original invention is based on an algorithm that has been published in the literature, but unfortunately leaves out key details that make it not possible for anyone else to implement a version that is literally compatible. That may not matter if you are doing all of your modeling with software from a single vendor, but if you want to mix and match, fingerprints generated by one package cannot be compared to fingerprints generated by another, even if the input molecules are the same and the implementation follows the same basic recipe: the numbers will be completely different. Because the CDK project previously did not have its own implementation, we have filled this particular hole. Anyone using software in a Java runtime environment can have access to it without having to pay anyone or ask for permission. We have put in a significant amount of elbow-grease to make sure that these fingerprints pass various validation tests, and perform with an enrichment rate comparable to other implementations. But perhaps more importantly, the algorithm has been very deliberately built in a way that is relatively easy to describe in words, and is based on code that is highly self contained. Definitions like implicit hydrogen count, aromaticity, ring blocks and chirality are minimalistic, well defined, and guaranteed never to change. This means that if you generate a list of fingerprints for a structure, you can store them in a database, and use them forever; you don’t need to version them and make sure they get rebuilt whenever one of the dependencies changes (which is a major headache with many software packages). And because the implementation is quite platform agnostic, a single source file can be translated line-by-line into a different development environment. In practice, you can use the CDK implementation to generate sample results, to make sure that the transplanted version is operating identically. As mentioned previously, this has already been done and is in use by the
TB Mobile app. We intend to explicitly document the algorithm in the scientific literature in the near future, to complement the freely available source code, but you will have to wait for that. In the meanwhile, if you feel brave, look for the file
CircularFingerprint.java in the CDK source, under the fingerprints hierarchy. This is also the first time I have actively worked with the CDK codebase. The project appears to be in the midst of a major overhaul, so it will be interesting to see what comes out the other end. Besides an important new class of fingerprints, that is!