

May 9, 2017
- From the desk of Peter Lind, CDD Advocate -
Machine learning
Machine learning is about designing and running computer programs which automatically improve with experience. A learning machine is supposed to do the right thing in response to input data, and it should have the ability to improve and do better as it collects feedback on how good its responses are. In formal studies we say that the machine has a task (T) which is executed with some performance (P), to be improved as the program gains experience (E).
Here are some examples of tasks:
-
- Make the right moves in a chess game, with the goal to win.
- Guess which items a customer is likely to buy.
- Predict tomorrow’s weather at a location.
- Control arm and leg movements of a walking robot.
- Predict metabolic fate of an envisioned drug molecule.
- Determine which email messages are spam.
Tasks can be very different in nature, but the common theme is that the task should be executed differently and appropriately in response to different input data. The input data can be a chess board position or it can be records of a customer’s previous purchases and historical browse behaviour. Execution of a task always involves production of output data, which can in itself be a result which has interest, or it can be a prescription of further action towards an end result. The learning process tries to tweak how tasks are executed for better performance, meaning that the result of a certain input is expected to change and improve as the machine gains experience.
Deep and shallow machine learning
If the computer executes its task in a one-step fashion where input data are transformed to output data directly in a straightforward process, then the learning machine is said to have a shallow architecture. If task execution is step-wise, such that the output of a first process is the input to a second process and so on, then we say that we have a deep architecture.
As an example of a shallow learning machine, consider a primitive spam filter algorithm which seeks to learn a spam detection rule based on the presence of spam indicating keywords. Perhaps the machine learns that the words ‘urgent’, ‘amazing’, ‘free’ and a few more are indicative of spam. Summing up a score based on the presence of these words is a simple process which is shallow, because building the sum is a single logical step.
As an example of a deep learning machine, consider an image-analysis system having the task to detect and classify objects in a digital photograph. The machine gets its input in a pixel format, and it needs to determine if one or more sets of pixels somewhere in the picture represent a certain object, such as a dog or a car. There is no way to go directly from pixels to classified object in a single step. The same object can take up different amount of space on the image depending on how close it was to the camera, and the pixel pattern will depend on the angle from which the photograph was taken. Objects may be partially obscured by other objects and their appearance in the image will depend on shadows. Also the same object type may come in different sizes and colors.
Solving the problem requires that input data are processed in steps where perhaps the first step finds contours, the second step works out 2D shapes enclosed by contours, a third step works on a yet higher level and so on until we arrive at object classes. The machine will need to learn how to perform each step well in order to get good at the overall object recognition task. This is deep learning.
Deepness is a quality of learning machine architectures, and we can have more or less of it, but there is no clear-cut limit between shallow and deep. This is because processes and steps can be defined in different ways.
Feature transformation
As described above, the deep learning machine processes raw features of the input data, such as pixels, and converts them into higher order features which have better expressive power in the domain of the end problem, such as 3D objects. These conversions are called feature transformations, and the machine will internally work with multiple levels of representation.
A designer of a learning machine need not know in advance what sort of feature transformations are needed to solve a certain problem. A machine can, if it has a sufficient number of training examples, work out what feature transformations are effective to performing its task.
Features may not correspond to human concepts and we may be unable to explain, in reasoning terms, how the machine arrives at a certain result.
In summary, feature transformation explains why deep learning is effective. A deep learning machine designer needs to understand how to allow for feature learning and feature transformation to happen, but he or she need not have any ideas in advance about the nature of intermediate features to be involved.
Biology
Interestingly, there is evidence that the brain uses a step-wise process and a form of feature transformation when processing and acting on visual data. (1)
Nerve signals travel from the eye to an area of the brain called the primary visual cortex. Signals originating from neighboring areas in the retina will stimulate neighboring cells in the first layer of the visual cortex. It appears that the primary cortex deals with low level features such as edges between areas of different brightness and color. Signals from the visual cortex are subsequently sent further on to areas where objects are recognized, and from there to areas with other responsibilities, such as analysis of motion.
This may be a curious fact for the data scientist, and some of early machine learning research has been inspired by results from neuroscience. But most developments today are based on results and theories from computer science and statistical learning, and there is only very rarely any intention to imitate how the brain works and learns.
One of the technologies of machine learning is called artificial neural networks or just neural networks. This technology is called so because a diagram explaining its workings looks like a cartoon drawing of networked biological neurons. Again, there is usually no intention to simulate a biological process.
Feedforward Neural Networks
There are many types of learning machines and many versions of each. We will briefly describe here a basic example of the type called feedforward neural networks. (2) There exists many advanced elaborations which are used in programs such as AlphaGo (3) and the latest version of Google Translate (4).
Below is an illustration of a feedforward network. The circles represent artificial neurons. At the bottom is a layer of input neurons and at the top is a layer of output neurons. The neuron layers between are called hidden layers. A shallow network has few hidden layers and a deep network has many. The hidden neurons are called so because they are internal to the learning machine and the world outside will not see them. The illustration shows an example with only thirteen neurons while a real program often has many thousands of neurons. The number of neurons in the hidden layers can vary.
Remember that the machine will perform tasks (T) with some performance (P) which should improve as it gains experience (E). We can use sets of numbers to encode both input and output, so for a machine a task is always about production of output numbers in response to input numbers.
When attempting a task, the machine will be in a feed forward phase. Input data are assigned to input neurons, which means that each neuron is assigned a numeric value. If the input data is from a black and white image, then we need as many input neurons as we have pixels in the image. Each input neuron is connected to a number of neurons in the first hidden layer. A connection means that during feed forward the value of a lower neuron will by multiplied with a weight factor (w) specific to the connection, and the product will be added to the neuron in the higher end of the connection. A mathematical function called the activation function is then applied to the sum to form the new value of the neuron. This goes on, layer by layer, until numbers have added up on the output layer. The connection-specific weights will be adjusted as the learning goes on. The weights are typically set to random values before we start learning, which means that we will get random results and useless performance the first time we try on some tasks.
Perhaps we have decided that the first output neuron should code for the probability that there is a cat somewhere in the input image. High output values will mean high cat probabilities, and vice versa. The machine will record the differences between actual and correct results after the feed forward. It then enters a weight update phase. A certain algorithm called backpropagation is used to update the weights of the network such that errors would be less if the same task should be issued again. As the name implies, backpropagation works from the output layer and back through the hidden layers. Learning with new tasks goes on as long as resources allow, or until performance no longer increases. Very often the same set of tasks is reused many times, and we then term each cycle of tasks a training epoch. The backpropagation algorithm, which is based on results from calculus, was proposed as a training method for neural networks by Stanford mathematical psychologist David Rumelhart in 1986. (5)
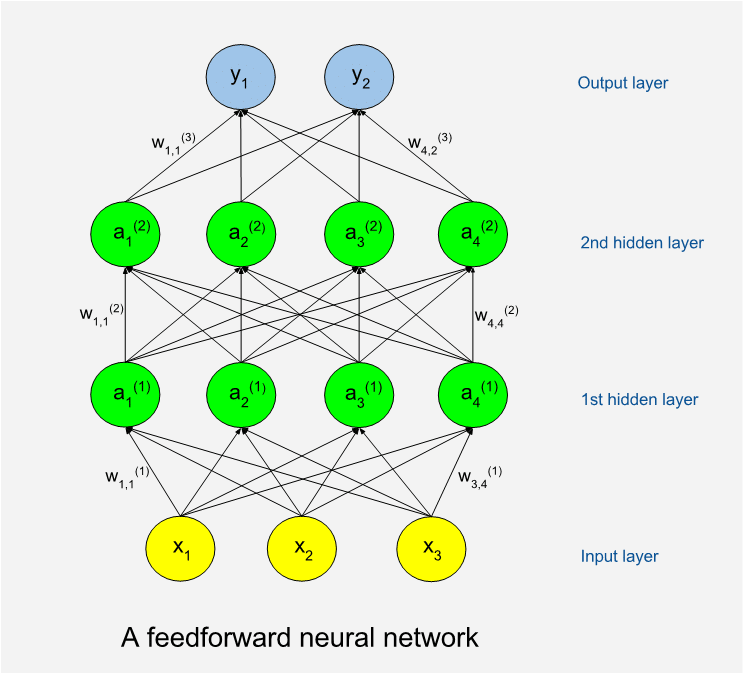
It was obvious from the start that networks could have more than a few layers, but training deep networks tends to consume more computer resources and much more training data are needed. This explains why deep learning has become relatively more important over time. In the last few years there has been a surge in popularity of deep networks not only because of more powerful hardware, but also because many more sufficiently big data sets have become available. A rough rule of thumb states that around 5000 training examples are needed for acceptable performance. (2)
Other Deep machines
One of the most legendary applications of machine learning is the chess computer Deep Blue by IBM. This is famous for having defeated grandmaster Garry Kasparov in 1997, who was the world champion at that time. The name Deep Blue has nothing to do with multi-layered networks and it was chosen for other reasons. (6) Blue is a main color of IBM’s brand identity, and deep was from the name of predecessor chess computer Deep Thought. The Deep Thought machine had got its name from a computer of the comedy science fiction series The Hitchhiker's Guide to the Galaxy, written by Douglas Adams. (7) Deep Thought of the fiction series was created with the purpose to compute the answer to the great question about Life, the Universe, and Everything. After thinking over the problem for seven and a half million years Deep Thought finally provided the answer, which was forty-two.
Deep Fritz and Deep Junior are other chess computers named in the same vein.
When it comes to computers and board games, most of the attention today is on the strategy game Go, which is more complex than chess as there are more alternatives per move. Go playing machines have only recently been able to defeat human grandmasters. A breakthrough came in 2016 when computer program AlphaGo defeated 9-dan master Lee Sedol. AlphaGo does indeed use a deep neural network. (3)
Deep learning in drug discovery
The desired effect of a drug is a result from its interaction with some biological target molecule in the body. Intermolecular forces bind drug and target molecules together and events following this will have effect on a disease or condition. Therefore a drug discovery project looks for compounds which can bind strongly enough to a target molecule. But a drug can also bind to non-target molecules in the body, which can cause unwanted and possibly dangerous side effects which must be avoided. A drug's absorption from the gut as well as its metabolism and excretion also depend on intermolecular forces.
Unfortunately, we cannot examine a potential drug molecule in the computer to predict how it will interact with the relevant molecules in the body. One reason is that we are lacking a good general method for prediction of intermolecular forces.
Today most candidate drugs fail when tried on humans in the clinic. This makes the overall drug making process extremely costly and much of resources could be saved if we had better predictive methods.
QSAR
Drug discovery is an iterative process and there is an element of trial and error. New compounds are designed based on test data of compounds already made. One tries to find patterns between structure and activity, or structure and any other property, in what is called a quantitative structure-activity analysis, abbreviated QSAR. QSAR analysis tries to model how drug-target interactions depend on drug structure, but it does this in an indirect fashion, without making any attempts to explicitly model the physical phenomena behind intermolecular forces.
QSAR models can be more or less local or global. A global model is trained on a diverse set of compounds and can produce meaningful estimates for a wide range of compounds. The data sets for global models tend to be large. A local model is trained on a compound class of special interest and it can produce estimates for compounds within that class. Local models generally perform much better than global models on sets of similar compounds, and they are therefore used in lead optimization projects where work is focused to certain classes of structures.
Deep neural networks show advantages when data sets are very large. As an example, the winning entry in a 2012 computational chemistry competition was using an ensemble of methods which included deep neural nets. (8) Data sets in the competition ranged from about 2000 to 50000 compounds and several thousands of descriptors were provided for each compound. Best results in this work were from networks with four hidden layers having in between 1000 to 4000 neurons in each of these layers. The mean R-squared statistics on the models was 0.49, meaning that these computer models can explain around half of the variance of data.
Another neural network QSAR study predicted compounds activities from multiple assays at the same time. (9) The study was on 19 data sets from PubChem ranging in size from about 2000 to 14000 compounds. Results were better than those from alternative methods, but changing the number of neural network hidden layers had no significant effect.
A drug discovery project will obviously want to be able to identify a candidate drug after synthesizing and examining as few compounds as possible, so the main interest in that context is for good models based on small data sets. Adding more layers to QSAR neural networks will most likely be of no advantage when data sets are small and the primary input consists of any of the traditional types of molecular descriptors.
Docking
Much effort has gone into developing so called docking programs which are algorithms with the purpose of predicting how well hypothetical molecules will bind to some target of interest. Docking programs can be used to screen a collection of virtual compounds to obtain a subset for which a higher proportion of compounds actually are good binders.
The docking program will need to examine a number of possible relative orientations between target and ligand molecules and estimate the binding strength for each such pose. A so-called scoring function is used for estimation of binding strength. The crux again is that we cannot today reliably predict how tight the interaction will be between two molecules.
Most programs dealing with molecular interaction will internally use a stick-and-ball type of representation of molecules. The models will also use factors expressing attractions, repulsions, flexibility and more. But being able to model the interactions between drug and target molecule with any level of precision is still a goal far away. Molecules in a biological environment move around, vibrate, and are surrounded by other molecules which affect the binding. A program which directly models the physical reality underlying molecular interaction would need to use a high level of theory and consider a number of relative poses and vibration modes which is so large that computation becomes unfeasible.
The classic stick and ball representation continues to be highly useful for prediction and explanation of many phenomena in chemistry, but it looks like computational chemists will need other features that do the work in intermolecular force models. The primary chemistry input will of course still be in the usual formats, but learning machines must be allowed to find suitable feature transformations.
A recent example of a machine learning study which uses deep learning for docking is by Pereira and co-workers. (10) The primary features used by their learning machine include context data for each atom of the compounds. Context data are distances, atom types, atomic partial charges and amino acids.
Synthetic methods
Another field where we can expect progress from machine learning is in retrosynthetic analysis. Molecules are often hard to synthesize and much of resources in drug discovery go into synthetic efforts. It can be difficult to come up with even a single synthetic route to a target compound. Retrosynthetic analysis is the systematic examination of possible synthetic routes, searched for in a backward manner, starting with a search for ways to make final compound from simpler compounds, then see how these can in turn be derived from even simpler compounds.
Again, availability of training data and collaborative efforts to collect data will be key for progress. An interesting and challenging aspect is that synthetic chemistry records are almost never totally complete as there is a practical limit to how much reaction result data a chemist can analyze and interpret. The learning systems will need to be good at handling incomplete data.
What will happen next?
Datasets
More and more data sets with chemical structures and activity data are released to the public, and there are now freely available databases providing both quality and quantity. (11) This is of enormous importance for the computing community which needs quality data in order to be able to develop new technologies. The general availability of benchmark data sets will continue to increase which will trigger accelerated developments of machine learning methods in drug discovery. It will seem increasingly odd to publish computational methods which are benchmarked on non-disclosed proprietary data sets.
Cloud
Pharmaceutical companies must protect their intellectual property carefully and therefore have very strict policies with regard to sharing of data. Fear that data will end up in the wrong hands has been the reason that the pharma industry was a late adopter of cloud computing. But Pharma is now externalizing more and more of their computational infrastructure. Providers of cloud based services are of course conscious about the security concerns of their customers and compete not just with technical service quality but also with security and trustability. Use of cloud services will continue to increase.
Software
A number of open-source frameworks for machine learning exists and a number of ecosystems for cloud computing have become standard tools for data scientists working with for example financial forecasting or customer behavior. It remains to be seen how R&D informaticians will integrate the standard tools of machine learning with the tools specific to the drug design domain. Members of R&D informatics staff are already expected to have a broad understanding about machine learning, and people from these fields, such as cheminformaticians, bioinformaticians, computational chemists and R&D IT staff, will continue to learn more about deep learning and exploit its possibilities.
Chemistry data for QSAR is of course just another form of data, like image data for object recognition, audio data for language interpreters, or board game positions for Chess or Go playing machines. Perhaps specific to QSAR is that real-life datasets are often small compared to those from other settings where often many orders of magnitude more samples are available. Deep learning requires large datasets. Perhaps deep networks can somehow be trained to find powerful feature transformations on large datasets with general data before they are further trained on smaller datasets of special interest.
Scoring functions
The lack of good technology for rapid estimation of intermolecular forces (scoring functions) is an area of particular interest. It exemplifies an area of technology where predictions about future progress have been over-optimistic for many decades. The use of deep learning methods may prove to be a way forward.
References
- https://en.wikipedia.org/wiki/Visual_system
- Goodfellow, I.; Bengo, Y. & Courville, A. (2016), ‘Deep Learning’, The MIT Press.
- Silver, D.; Huang, A.; Maddison, C. J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; Dieleman, S.; Grewe, D.; Nham, J.; Kalchbrenner, N.; Sutskever, I.; Lillicrap, T.; Leach, M.; Kavukcuoglu, K.; Graepel, T. & Hassabis, D.: ‘Mastering the game of Go with deep neural networks and tree search.’ Nature 529 (2016), Nr. 7587, 484–489
- Wu, Y.; Schuster, M.; Chen, Z.; Le, Q. V.; Norouzi, M.; Macherey, W.; Krikun, M.; Cao, Y.; Gao, Q.; Macherey, K.; Klingner, J.; Shah, A.; Johnson, M.; Liu, X.; Łukasz Kaiser; Gouws, S.; Kato, Y.; Kudo, T.; Kazawa, H.; Stevens, K.; Kurian, G.; Patil, N.; Wang, W.; Young, C.; Smith, J.; Riesa, J.; Rudnick, A.; Vinyals, O.; Corrado, G.; Hughes, M. & Dean, J.: ‘Google's Neural Machine Translation System: Bridging the Gap between Human and Machine Translation.’ arXiv(1609.08144v2).
- Rumelhart, D. E.; Hinton, G. E.; Williams, R. J.: ‘Learning representations by back-propagating errors.’ Nature 323 (1986), Nr. 6088, S. 533–536
- Hsu, F.: ‘Behind Deep Blue: Building the Computer that Defeated the World Chess Champion’: Princeton University Press., 2002
- https://en.wikipedia.org/wiki/Deep_Thought_(chess_computer)
- Ma, J.; Sheridan, R. P.; Liaw, A.; Dahl, G. E. & Svetnik, V. (2015), 'Deep Neural Nets as a Method for Quantitative Structure-Activity Relationships', J. Chem. Inf. Model. 55(2), 263–274.
- Dahl, G., E.; Jaitly, N. & Salakhutdinov, R. (2014), 'Multi-task neural networks for QSAR predictions', arXiv(1406.1231).
- Pereira, J. C.; Caffarena, E. R. & dos Santos, C. N. (2016), 'Boosting Docking-Based Virtual Screening with Deep Learning', J. Chem. Inf. Model. 56(12), 2495–2506.
- Bento, A. P.; Gaulton, A.; Hersey, A.; Bellis, L. J.; Chambers, J.; Davies, M.; Krüger, F. A.; Light, Y.; Mak, L.; McGlinchey, S.; Nowotka, M.; Papadatos, G.; Santos, R. & Overington, J. P.: ‘The ChEMBL bioactivity database: an update’. In: Nucleic Acids Research 42 (2013), Nr. D1, S. D1083--D1090